Getting Started¶
So at this point you have already completely installed <install.html>_ miRPursuit. You should be familiar with the various stages <stages.html>_ in the pipeline and have step up all the necessary configuration files. You are now ready to run miRPursuit for the first time.
You can run with these default settings or you can start with your customized settings. MiRBase will be downloaded by the install script but you should have a genome file or download one to run (Try Phytozome or ensemble plants). For the purpose of this example all instructions will be based on the following configurations in Table 3.
Table 3 - Example configuration of workdirs.cfg
| Variable | Value |
|---|---|
| workdir | ${HOME}/miRtest/ |
| MEMORY | “4g” |
| THREADS | 2 |
| INSERTS_DIR | ${miRPursuit}/testDataset/ |
| GENOME | ${SOURCE_DATA}/Genome/Genome.fa |
| GENOME_MIRCAT | ${GENOME} |
| FILTER_SUF | 18_26_5 |
| ADAPTOR | “TGGAATTCTCGGGTGCCAAGG” |
Bash variables in table 3 and their values:
(Keep it simple, store all dependant DBs in $SOURCE_DATA it will be simpler to configure. But substitute appropriately.)
SOURCE_DATA=${HOME}/souce_data
(This is the path used in this example. Depending on where you stored your installation, you should substitute appropriately.)
miRPursuit=${HOME}/git/miRPursuit
(In this case we don't specify a user. In reality this would expand to your home dir. /home/[user])
HOME=/home/
How to run the program¶
Run the command:
${miRPursuit}/./miRPursuit.sh -f 1 -l 2 --fasta test_dataset-
- This is the simplest test case. Let’s break down this command.
- ${miRPursuit}/./miRPursuit.sh - This is used to execute the main script to start miRPursuit. You can simply run ./miRPursuit if you’re current path is already in the miRPursuit directory.
- -f - The number of the first library
- -l - The number of the last library
- –fasta test_dataset- - Run in fasta mode, and use all libraries that have the string “test_dataset-” preceding the sequential numbering.
Important
MiRPursuit is designed to run an interval of libraries. So it will run all libraries starting with test_dataset-1.fa (-f first in this example 1), up to test_dataset-1.fa (-l last in this example 2). The files to be processed in your INSERTS_DIR should have a common string along with a sequential numbering.
However you can also use it in specific file mode. That is specify the file you want to process for a single run.
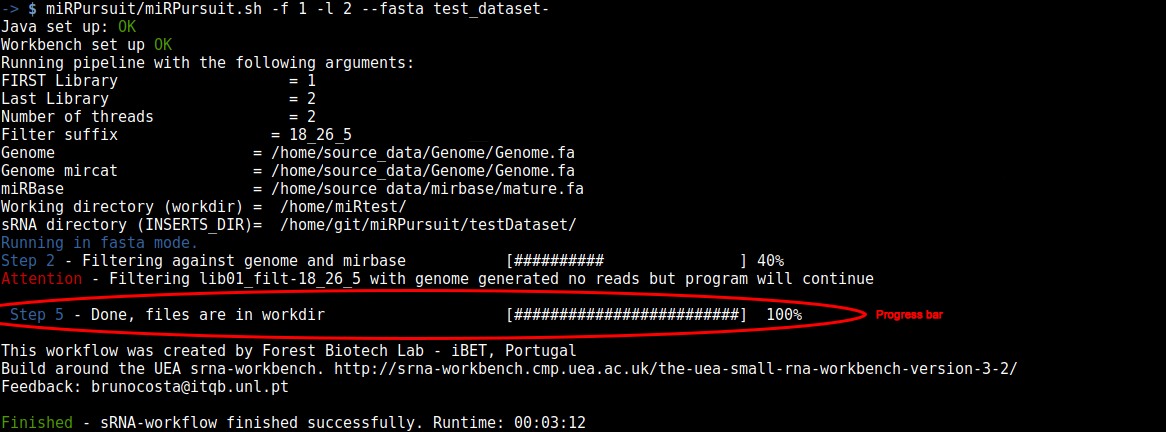
Figure 2 - miRPursuit full run in fasta mode for libraries 1 and 2.
In Figure 2, a complete run of miRPursuit is shown using the above specified command. The first procedure the pipeline does is to check that all variables are defined and their values link to existing files. If any of the mandatory files is missing a warning will be issued and you will have to start the run again (Fig. 3).
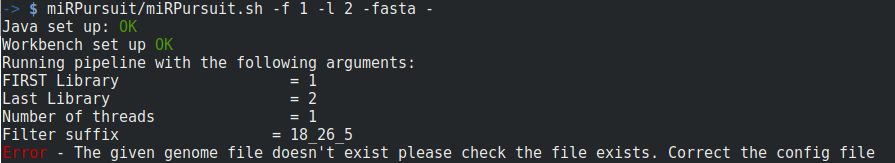
Figure 3 - Warning issued by miRPursuit due to lack of genome file.
During the miRPursuit run the progress bar will update you as to which stage is in execution, along with its percentage of conclusion. Once the run is finished a finished message will be given as shown in figure 2.
Important
Tip: In order to run on remote servers without the need to stay connected try screen
Run options¶
- The full listing of the options available
- -f|–lib-first First library to be processed.
- -l|–lib-last Last library to be processed.
- -h|–help See the list of options.
- Optional arguments
- –lib Set the library number that should be assigned to the specified file. (Only relevant if using specific files)
- –fasta Set the program to start using fasta files. As an argument supply the file name that identifies the series to be used. Ex: Lib_1.fa, Lib_2.fa, .. –> argument should be Lib_
- –fasta (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now).
- –fastq Set the program to start using fastq files. As an argument supply the file name that identifies the series to be used. Ex: Lib_1.fq, Lib_2.fq, .. –> argument should be Lib_ , if no .fq file is present but instead a .fastq.gz file will additionally be extracted automatically.
- –trim Set this flag to perform adaptor triming. No argument should be given. The adaptor is in the workdirs.cfg config file in the variable ADAPTOR.
- -s|–step Step is an optional argument used to jump steps to start the analysis from a different point
- Step 1: Adaptor trimming (If flagged) & Wbench Filter
- Step 2: Filter Genome & mirbase
- Step 3: Tasi
- Step 4: Mircat
- Step 5: Reporting
Specific file mode * –fasta${NC} (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now). * –fasta${NC} (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now). * –lib${NC} (Optional) (In specific mode. i.e. no -f and -l) Set the library number to be attributed to the file. Should be coupled with –fasta or –fastq.
Both fasta and fastq options work in the same manner they require the preceding string to the sequential numbering that all libraries have in common. Ex:
- Lib01.fa
- Lib02.fa
- Lib03.fa
The common string is “Lib” or “ib” or “b”, the sequential numbering is 01,02,03. And .fa is the extension.
Attention
Avoid using spaces in file names. As it might generate unexpected errors.
If the –trim flag is present in the command the reads are then searched for adaptor sequences using the fastx_clipper software of the FASTX toolkit; sRNA sequences are assumed to be the string of nucleotides between the 5’ and 3’ adaptor sequences.
Additionally if the –fastq option is used. A fastqc quality report will be generated for each of the libraries.